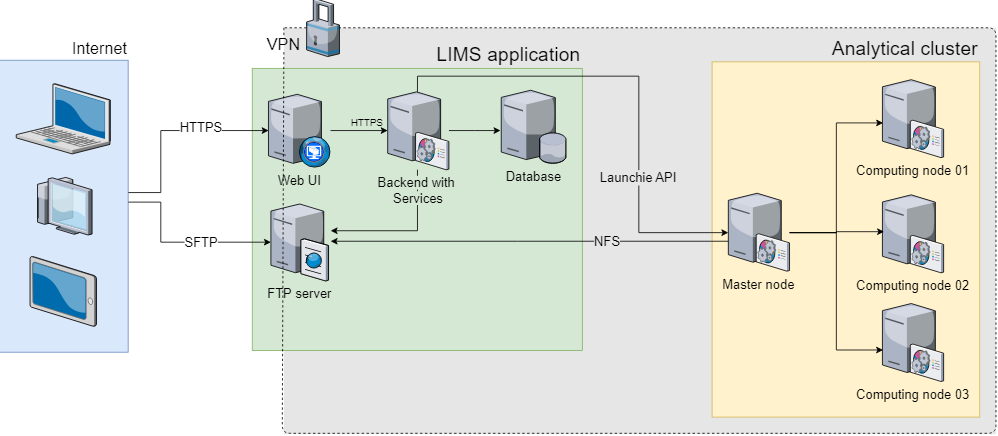
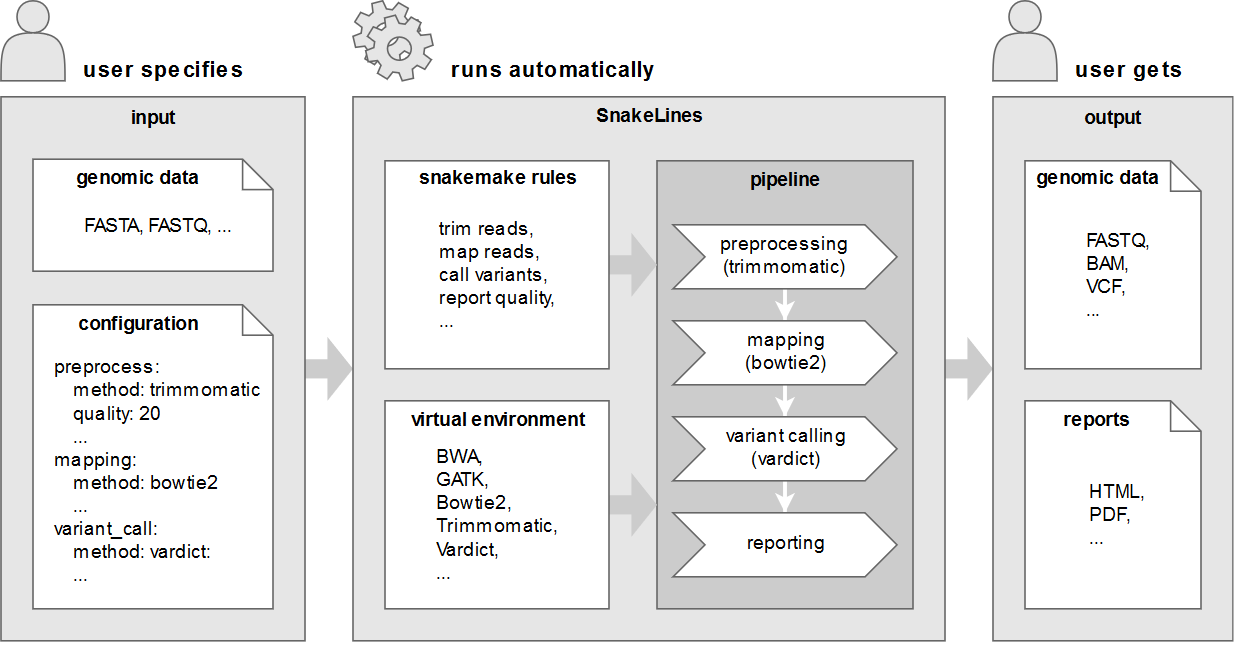
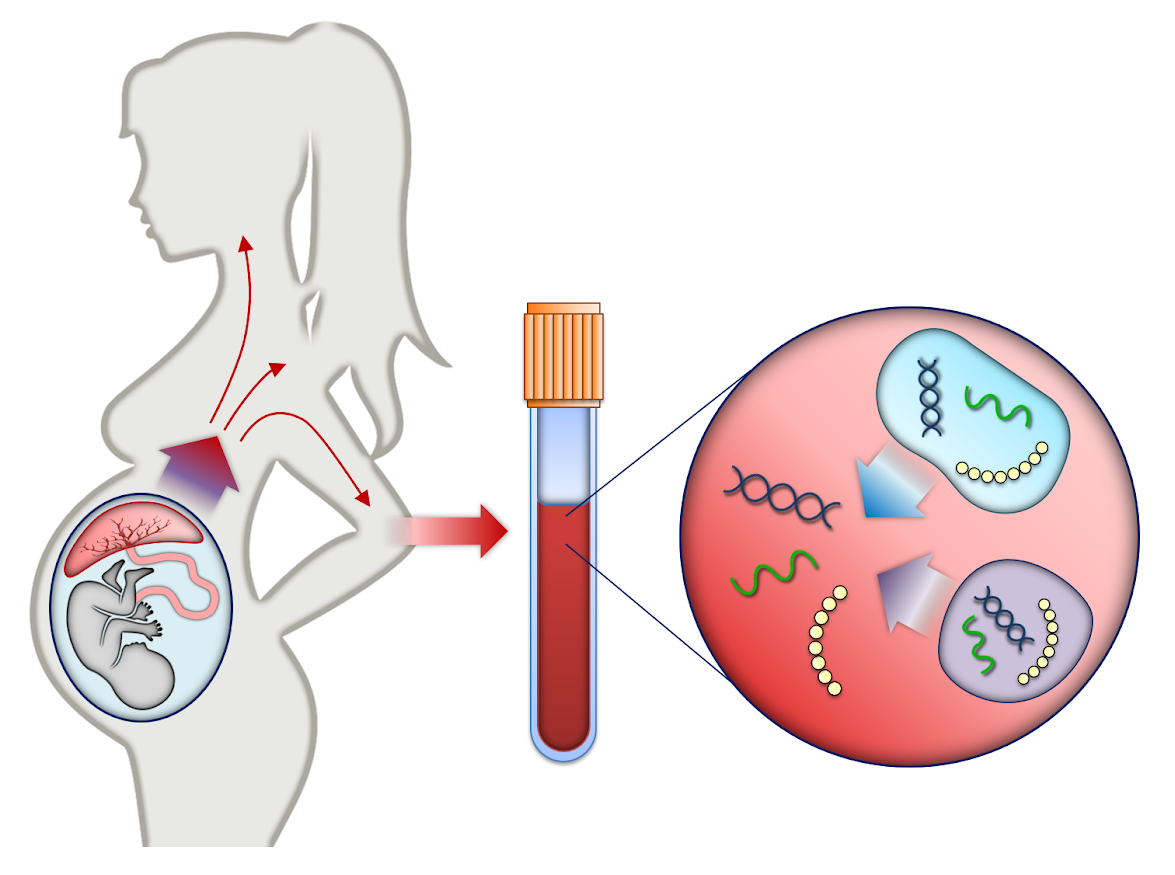
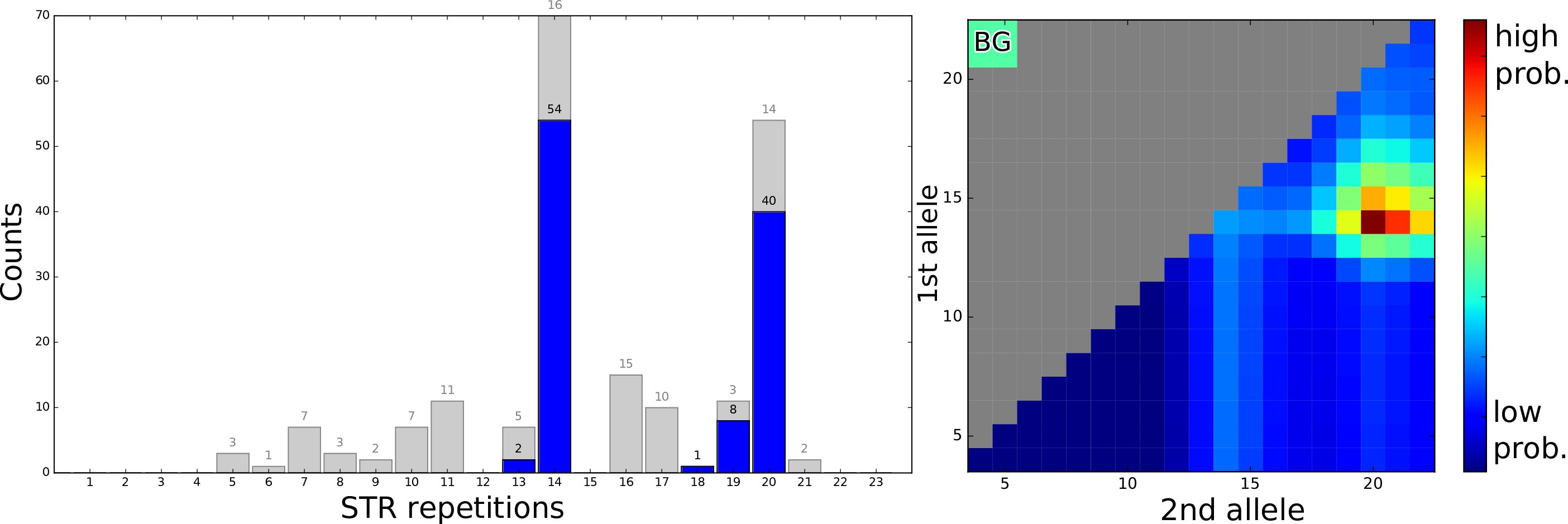
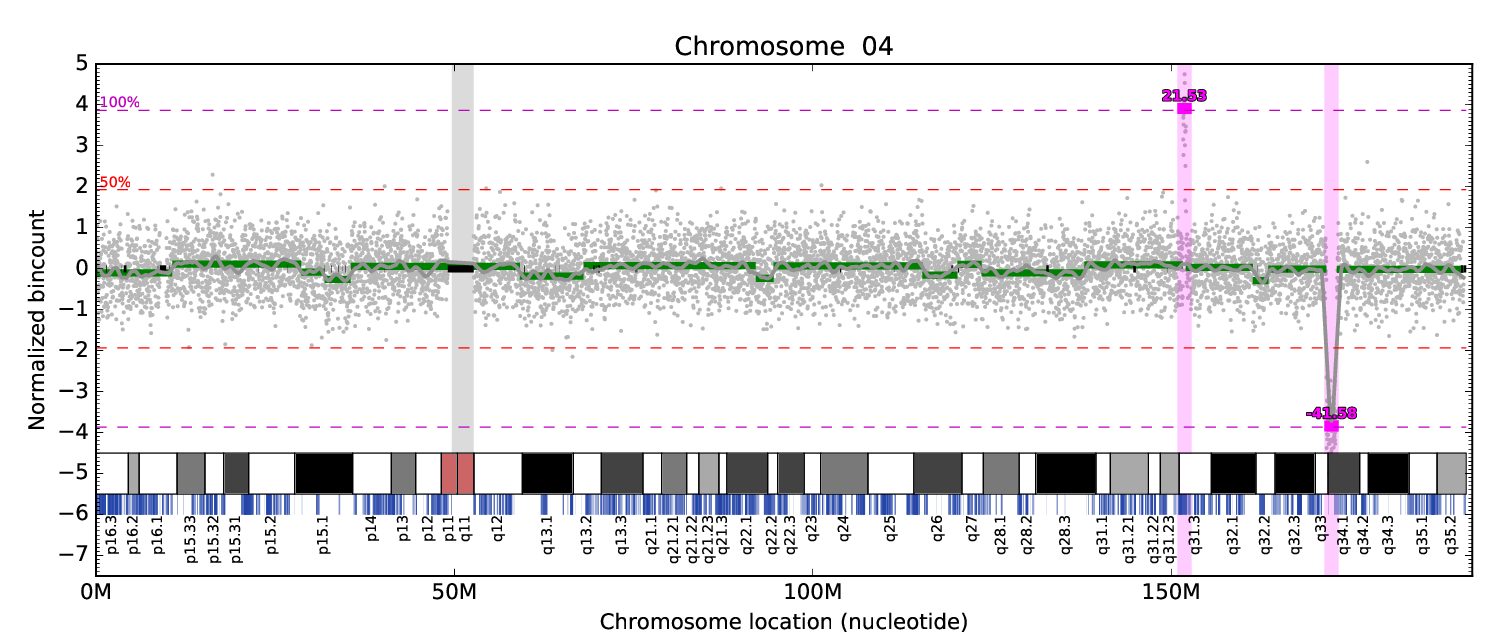
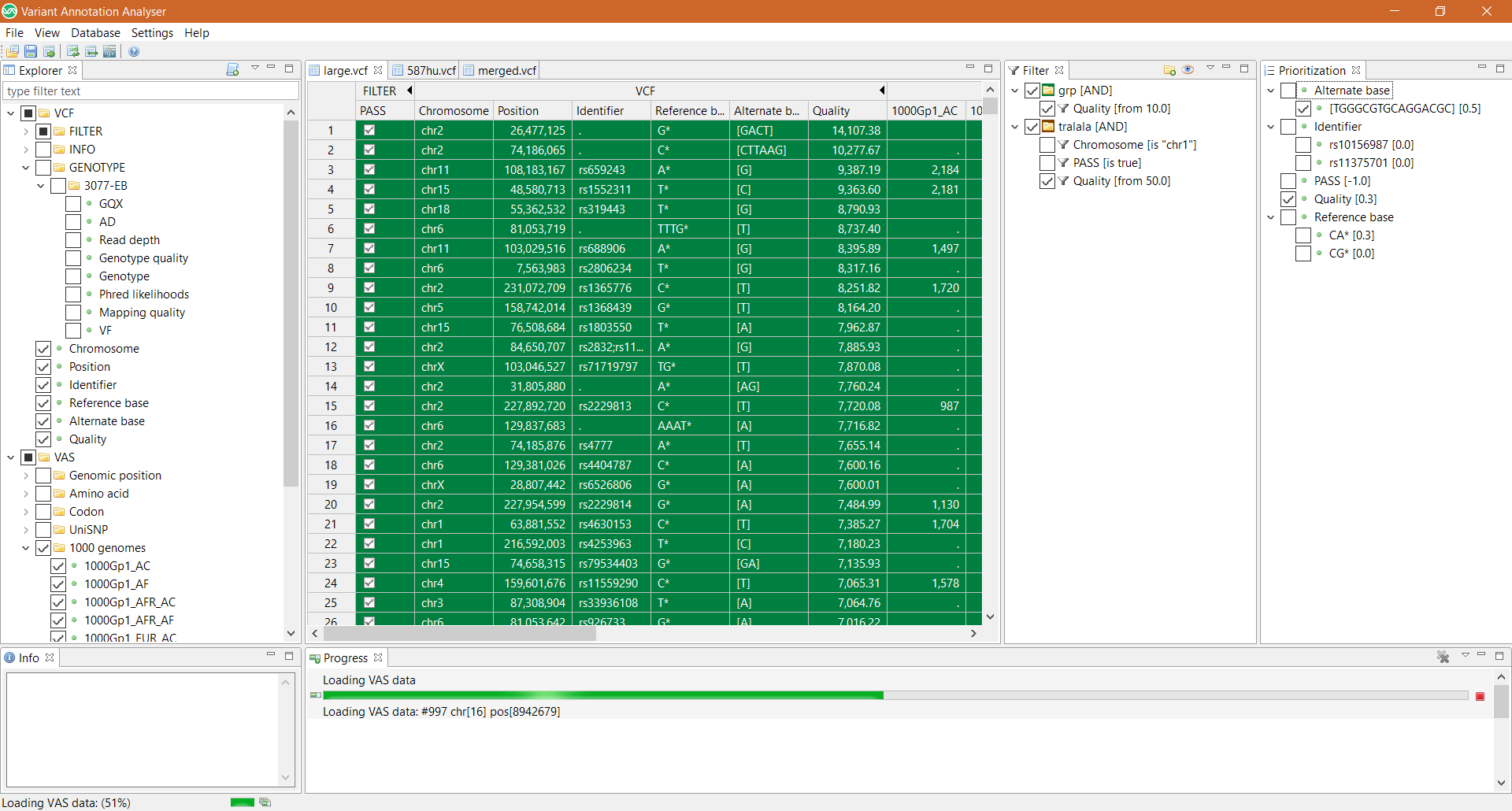
Väčšina multi-projektových výskumných laboratórií (ako sú zdielané laboratória) sa pokúša vysporiadať s registráciou, sledovaním, integráciou a monitorovaním vzoriek a ich analýz. Keďže tieto údaje obsahujú osobné informácie, zabezpečené úložisko a prístup má vysokú prioritu aj kvôli novým európskym právnym predpisom. Na zvládnutie týchto výziev sme vyvinuli webový systém riadenia informácií v laboratóriu (LIMS) optimalizovaný pre…
Read More