
Introduction
Analyses based on massively parallel sequencing detect vast amounts of variants, while only few are responsible for traits of interest. Successful identification of these few variants requires annotation with various attributes – especially function prediction score and conservation score. Different annotation data is scattered across various databases, which makes manual annotation a time-consuming and tedious process.
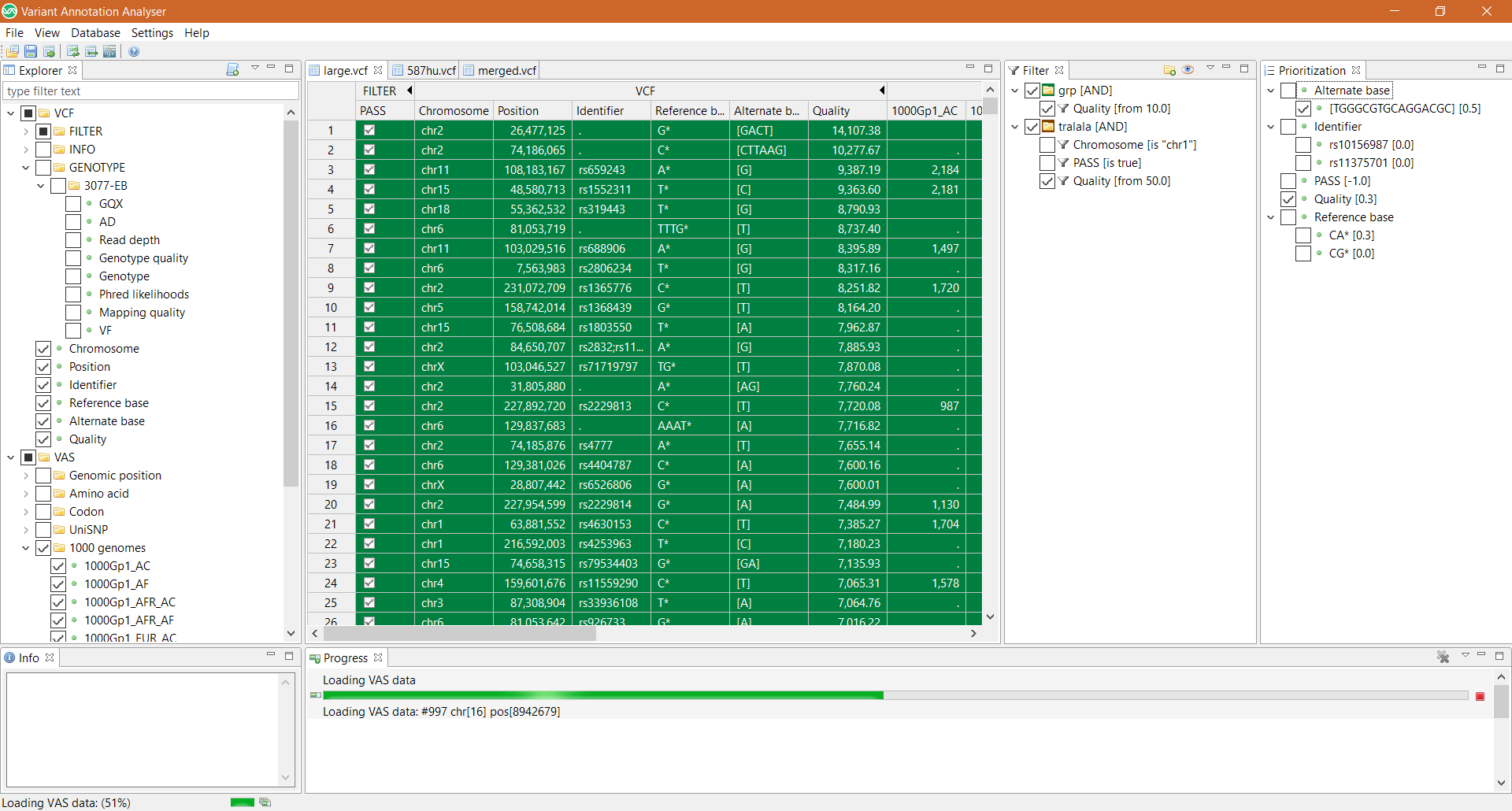
Variant Annotation Analyser
To facilitate variant annotation process, we developed a desktop application called Variant Annotation Analyzer (VAA) together with web based Variant Annotation Service (VAS). The VAA acts as thick client on VAS, which currently serves as it’s sole annotation provider.
To annotate variants user has to simply load VCF file into the application and select desired attributes. All visible VCF records at the moment are automatically annotated one by one in display order. Application also supports follow-up prioritization process by filtering and sorting operations which are specific for each attribute data type. Attribute filters are defined in Filter module and can be composed into filtering groups with specific meaning and logical rules. Prioritization module allows user to assign weights to desired set of attributes. Ordering score is than assigned to each variant.
Working data can be exported to xlsx, csv, tsv formats for further processing by an external spreadsheet editor. Moreover, modular programming architecture enables easy integration of new features and additional annotation providers.
Variant annotation Service
The service acts as a web interface for the database of non-synonymous SNP’s functional predictions – dbNSFP [1,2] and provides more then 100 attributes collected from various commonly used variant and gene databases, conservation scores and results of functional prediction tools. The use of the web service is not limited to the VAA application and would be fully opened to any academic use.
Machine learning prioritization
We have also developed machine learning prioritization process based on user preference which has yet to be implemented into the application. The purpose of this upcoming feature is to aid user by finding variants similar to his selection. Since user may not know all attributes that are relevant to his trait of interest, all significant attributes and their weights can be estimated by trained machine learning model. Prioritization module is supposed to import these weights and sort variants accordingly.
Conclusion
The VAA application and the VAS web service provide researchers with rich and automated annotation of variants in a fraction of time compared to manual annotation. Web based annotation relieves user from storing vast database locally. With the option of further prioritization it provides a powerful tool for fast identification of potential candidate mutations among loads of irrelevant variants.