
Introduction
Development of sequencing technologies with massive parallel throughput allowed laboratories around the world to analyze DNA fragments of various organisms. However, actual use of DNA sequencing data is impeded by two main obstructs. Firstly, DNA data processing and interpretation is often difficult for researchers and clinicians with small computational background. Secondly, due to the complexity of an available bioinformatic software, where one tool is dependent on specific version of another tool, reproducing data analyses in another computational clusters is often too troublesome.
Results
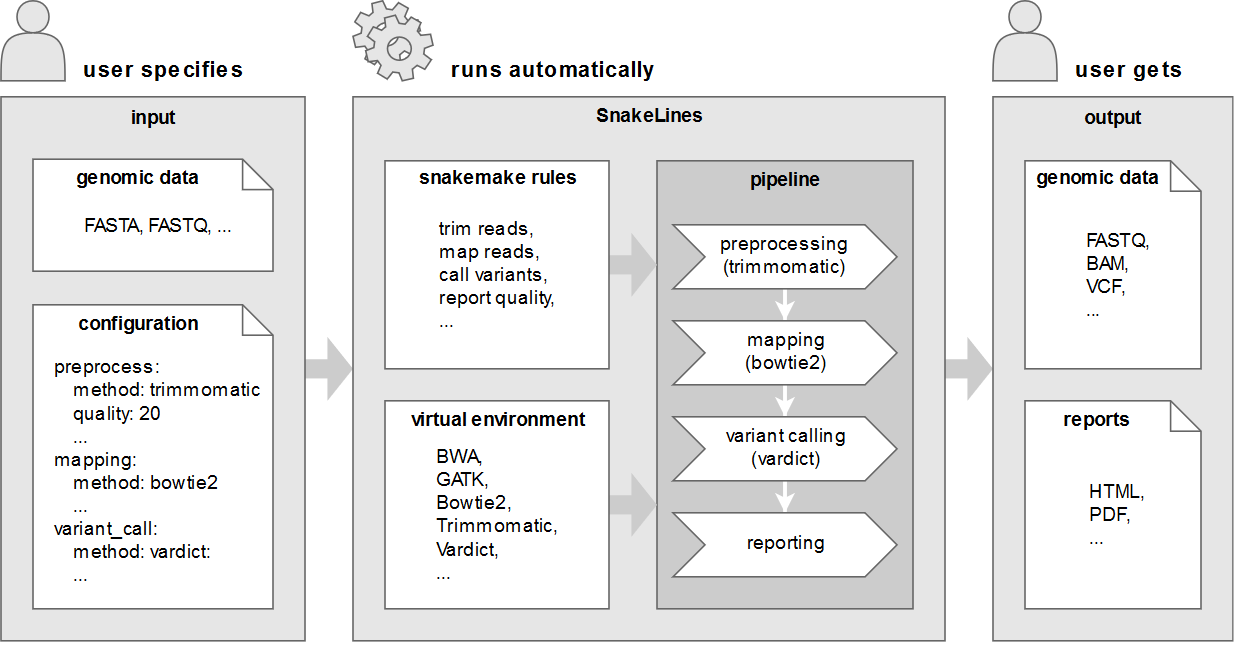
We propose a framework together with an implemented set of computational pipelines, called SnakeLines, for processing of paired-end Illumina reads, including mapping, assembly, variant calling, viral identification, transcriptomics, metagenomics, and methylation analysis. Our framework implements a self-created virtual environment that contains required tools and libraries and isolates them from host operating system, thus ensuring easy portability and reproducibility across different Unix-based systems.
Availability
The open-source code of the pipelines, together with test data,
is freely available for non-commercial use from https://github.com/jbudis/snakelines.